Tools & Techniques
Sexy sequencing
Just So Stories: How we got seduced into whole-genome sequencing
The days when PhDs were minted based on sequencing a few nucleotides at the end of a piece of nucleic acid are definitely gone. The countless hours spent doing Maxam-Gilbert sequencing and reading gel autoradiographs by hand have gone too. Sixteen-lane ABI Sanger dideoxy sequencing machines have now been replaced by ultra-high throughput short-read sequencing. Whole-genome sequencing now rules.
But what is so seductive about whole-genome sequencing?
Is it the fact that the $1,000 whole-genome sequence is now a reality?
Is it the technological beauty of whole-genome sequencing?
Is it what the whole-genome sequence actually tells us?
The answer is probably a mixture of all three.
There is no question that walking into a room full of HiSeq X Ten short-read sequencing machines (which Illumina Inc. sells in batches of 10) is an impressive sight. Equally impressive, but in a different way, is the long-read sequencing machine, from Pacific Biosciences of California Inc. (PACBIO), which can sequence up to 10,000 consecutive base pairs at a time on a good day. Now there is nanopore sequencing, which is done on an instrument not much bigger than a USB flash drive, and companies such as Oxford Nanopore Technologies Ltd. are starting to make an impact, with the sequencing of bacterial and virus genomes such as Ebola (see Figure: A sequence in time).
Sexy is a good start. But what has and can the technology deliver?
For babies born with a developmental abnormality, sequencing of the parents' and child's DNA will sometimes give a diagnosis of what is wrong, and can lead to clinical interventions that potentially could allow relatively normal lives for the whole family. Several neurodevelopmental pediatric epilepsies fall into this category.
Comprehensive sequencing is a cost-effective way to proceed, because the alternative is to check different known genes one by one in a process of elimination, which takes much longer and costs more money than doing it all in one step.
In oncology, diagnostic gene panels are being used to look for mutations in defined genes. Good examples include mutations in the EGFR in non-smokers with lung cancer, KRAS mutations in colon cancer, or chromosomal translocations such as EML4-ALK and the like in lung and other cancers.
These diagnostic tests are used routinely but are particularly useful in lung cancer, given that there are a number of drugs available for patients who have particular mutations in their tumor cells, such as AstraZeneca plc's Iressa gefitinib for EGFR mutations or Pfizer Inc.'s Xalkori crizotinib for EML4-ALK translocations.
Exome sequencing is also being used to search for coding mutations in tumor genomes, with the same clinical intent - to find mutations in genes controlling proliferation of the tumor cells that can be used to direct clinical treatment.
In immuno-oncology the biggest issue is going to be recognizing responder and non-responder patients for the expensive checkpoint inhibitor antibodies, such as those that block CTLA4, PD-1 or PD-L1. Exome sequencing is being used to catalogue neo-antigens, which can then be reviewed in the context of the patient's MHC. That's an important component of this new paradigm because the ability to present the neo-antigens is connected to the immune system's ability to respond once the checkpoints are released.
The recognition that anti-EGFR antibodies only worked in KRAS wild-type tumors and not in those with KRAS mutations was a profoundly instructive example of how to save money and avoid expensive treatments that do not work.
Mutations in germline DNA are also important. For example, changes in homologous repair genes such as BRCA1 or PTEN would indicate that PARP inhibitors be used, as those drugs clearly are more efficacious in tumors where homologous repair pathways are not working. As has been demonstrated for the treatment of HIV, the name of the game in oncology is to identify and keep ahead of the mutations in the proteins that provide resistance to the available drugs.
New paradigm, new paradox
Whole-genome sequencing will in time become the technology of choice in clinical practice, but the limitation is not the ability to uncover mutations; it's the availability of drugs tailored to altered proteins.
Nevertheless, if the first thing analyzed from a whole-genome sequence is obvious mutations in coding regions - then why not just do exome sequencing, which appears on the face of it to be cheaper and easier?
The answer is that copy number variants, repeats, insertions and deletions in genomic DNA will be missed by exome sequencing.
Despite the fact that for a subset of these genomic changes we know what the consequences are or might be, there are a large number of simple and complex sequence variants that exist where currently we have little or no idea what they mean.
It is also time-consuming and not cost-free to do whole-genome sequence analyses in a background of widespread variation. The cost of sequence analysis is not quite such a dark secret as it was, but it does make the "$1,000 whole-genome sequence" still a little misleading. High-quality whole-genome sequencing - maybe using more than just short-read methods - might cost you more, but that quality will save you analysis time, and therefore money in the end.
Consistent precision
While everyone's hopping on the bandwagon of whole-genome sequencing to push the precision medicine envelope, it's easy to anticipate some problems.
Many of the precision medicine initiatives of different governments, including the president's Precision Medicine Initiative, Genomics England, or programs in Estonia and Asia, are calling for comprehensive whole-genome sequencing so that association studies can be done against a large amount of clinical phenotype data.
This is all very well in principle, but as the U.K. and others are finding out the logistics are far from trivial.
In any association genetics study consistency in clinical phenotyping (i.e., collecting the same information in the same way for each patient) is a prerequisite. Most genome-wide association studies have been successful because the phenotypes are simple and robust - disease or no disease.
It gets increasingly difficult to find associated common alleles to complex phenotypes because as the phenotyping complexity goes up, the consistency goes down, along with the number of people with the particular phenotype in question. This reduces the power of the study to find associated alleles very rapidly.
So the information generated is only as good as the phenotypes against which the sequence variation is measured, which means that great care must be taken to ensure consistency of phenotype. Most electronic medical records that are being used as a place to capture phenotypes are probably not robust enough for this yet but can be adapted to meet that standard.
A clear purpose
We need to be clear about why we are doing these large studies.
Is it to find common and rare alleles that predispose to disease? To find alleles that are causative and as such point to potential drug targets? To subtype disease into molecular categories, as is being done in different cancers? Or is it to stratify patient populations for clinical management or clinical trials?
These are all important questions. But the component that should not be underemphasized is that we need to discover the drugs to treat the genetically and phenotypically defined populations in a "precise" way, so the drugs work in the majority of the patients for whom they are prescribed.
Encouraging the commercial side of the business to embrace this approach comprehensively is a necessary prerequisite.
Fortunately for patients, most pharmaceutical and biotech companies have got the message. As well as being involved in global precision medicine initiatives, they are sponsoring related studies using more genetically isolated or more accessible populations of patients in places like Finland to answer these questions.
Other companies have also sponsored their own large-scale exome-sequencing projects in concert with some of the payers (such as the partnership between Regeneron Pharmaceuticals Inc. and Geisinger Health System) to look for rare predisposing mutations to common polygenic diseases. Even larger projects are under discussion.
It is easy to be seduced by technology as well as one's own hyperbole, and whole-genome sequencing is rife with both seduction and hyperbole. But sooner or later we will all have our genomes sequenced and we will know what the sequences mean.
Then the dinner table or coffee time conversation will not be: "Have you done 23andMe or Ancestry.com" and "Do you metabolize asparagus in a particular way?" Or even more interestingly, "I have some exotic genes in my DNA - I wonder where they came from…"
Au contraire, it will be a much more definitive statement of your inheritance and what drugs are right for you, if and when you get the diseases that you are predisposed to. This is how it will be.
Companies and Institutions Mentioned
AstraZeneca plc (LSE:AZN; NYSE:AZN), London, U.K.
Geisinger Health System, Danville, Pa.
Illumina Inc. (NASDAQ:ILMN), San Diego, Calif.
Oxford Nanopore Technologies Ltd., Oxford, U.K.
Pacific Biosciences of California Inc. (NASDAQ:PACB), Menlo Park, Calif.
Pfizer Inc. (NYSE:PFE), New York, N.Y.
Regeneron Pharmaceuticals Inc. (NASDAQ:REGN), Tarrytown, N.Y.
23andMe Inc., Mountain View, Calif.
Targets and Compounds
BRCA1 - Breast cancer 1 early onset
CTLA4 (CD152) - Cytotoxic T-lymphocyte associated protein 4
EGFR - Epidermal growth factor receptor
EML4-ALK - EML4-ALK oncogene fusion protein
KRAS - K-Ras
MHC - Major histocompatibility complex
PARP - Poly(ADP-ribose) polymerase
PD-1 (PDCD1; CD279) - Programmed cell death 1
PD-L1 (B7-H1; CD274) - Programmed cell death 1 ligand 1
PTEN (MMAC1; TEP1) - Phosphatase and tensin homolog deleted on chromosome ten
Figures
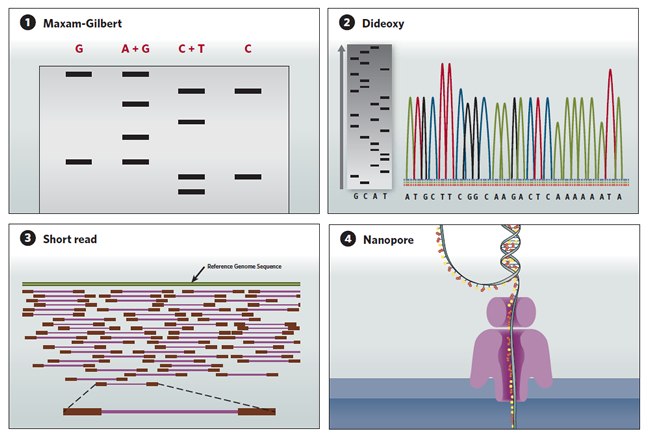
A sequence in time
DNA sequencing technologies have evolved since the Maxam-Gilbert technique of the 1970s, to today's automated high throughput methods.
(1) Maxam-Gilbert sequencing:DNA is radiolabeled and split into aliquots, then subjected to one of four chemical modifications that affect either adenine and guanine (A+G), guanine alone (G), cytosine and thymine (C+T), or cytosine alone (C). The chemicals are titrated to induce about one modification per DNA strand, and the DNA is then cleaved adjacent to the modifications. The degraded products are size-separated on a polyacrylamide gel and autoradiography is used to visualize the fragments. The sequence is then inferred by the presence or absence of breaks induced by the chemical modifications.
(2) Sanger dideoxy sequencing:In this sequencing-by-synthesis method, the DNA is split into four aliquots and mixed with DNA polymerase, a labeled primer that initiates synthesis, normal deoxynucleotides that are incorporated into the newly synthesized chain, and a low concentration of dideoxynucleotide versions of either A, C, G or T, which terminate the sequence. The synthesized fragments are then size-separated on a polyacrylamide gel, and the sequence is read (gray gel). A later version of the technology uses fluorescently labeled dideoxynucleotides run on capillary gels, which are detected electronically (multicolored peaks).
(3) Short-read sequencing:Next-generation high throughput sequencing technologies have parallelized the sequencing-by-synthesis process. The DNA to be sequenced is cut into fragments containing a few hundred nucleotide base pairs (purple) and labeled on both ends with adaptor sequences (red). The adaptors hybridize to primers immobilized onto a bead or flow cell. After PCR amplification of each fragment, a sequencing primer is added and, one at a time, either A, C, G or T is flowed through the reaction chamber. If the nucleotide in the chamber pairs with the next nucleotide on the template fragment, the pyrophosphate released during nucleotide incorporation triggers an enzymatic reaction that releases a flash of light, allowing the nucleotide incorporated to be determined. The short reads obtained from each fragment are then assembled and compared to a reference genome sequence (green).
Long-read sequencing(not shown): Pacific Biosciences of California Inc. (NASDAQ:PACB) developed a single-molecule real-time (SMRT) sequencing platform that allows sequencing of tens of thousands of nucleotides per read. In microscopic wells that allow limited light to enter through the bottom, individual DNA strands are sequenced by synthesizing complementary strands. The complementary strands are synthesized using nucleotides that are distinguished by fluorophores conjugated to their terminal phosphates. When a nucleotide is incorporated, its fluorescent phosphate is released, and different emissions are detected according to the different nucleotides in the sequence. Because only a very small volume at the bottom of the well is illuminated, the fluorophore diffuses away from the illumination zone once released, allowing sequencing of a large number of nucleotides without interference.
(4) Nanopore sequencing: Instead of relying on DNA cleavage or synthesis to infer DNA sequences, the nanopore sequencing method directly detects the molecular properties of different nucleotides. A single-stranded DNA molecule is extruded through a protein nanopore embedded in a membrane, and an ion current is run through the pore. As the DNA strand passes through the nanopore, the identity of the nucleotides can be inferred by analyzing the amplitude and duration of transient blocks in the current. <img height="435" src="innovations20162q16toolsImage04071605.jpg" width="650"/>